Article Text

Abstract
The primary goal of this article is to critically discuss the syndromic overlap that exists between early behavioural variant frontotemporal dementia (bvFTD)—the most common clinical syndrome associated with frontotemporal lobar degeneration (FTLD)—and several primary psychiatric disorders. We begin by summarising the current state of knowledge regarding FTLD, including the recent discovery of FTLD-causative genetic mutations. Clinicopathological correlations in FTLD are subsequently discussed, while emphasising that clinical syndromes of FTD are dictated by the distribution of FTLD pathology in the brain. We then review a large number of cases with suspected and confirmed bvFTD that had previously been diagnosed with a primary psychiatric disorder. The clinical and neuroscientific implications of this overlap are discussed, focusing on the importance of early diagnosis for clinical and therapeutic reasons. We propose that largely due to the paucity of biomarkers for primary psychiatric disorders, and the limited use of FTLD-related biomarkers by psychiatrists at present, it is very difficult to separate patients with early bvFTD from those with primary psychiatric disorders based on clinical grounds. Furthermore, specific limitations of the Diagnostic and Statistical Manual of Mental Disorders (DSM) 5 criteria for bvFTD may inadvertently discourage recognition of bvFTD in mental health settings. Clinically, more research is needed to develop tools that allow early differentiation of bvFTD from primary psychiatric disease, as bvFTD therapies will likely be most effective in the earliest stages of disease. From a neuroscience perspective, we argue that bvFTD provides an excellent paradigm for investigating the neural basis of psychiatric disorders.
- DEMENTIA
- FRONTAL LOBE
- NEUROPSYCHIATRY
- BEHAVIOURAL DISORDER
- PSYCHIATRY
Statistics from Altmetric.com
Introduction
The term frontotemporal lobar degeneration (FTLD) is a neuropathological designation used to identify a group of neurodegenerative diseases of the frontal and anterior temporal lobes (ATLs) of the brain caused by non-Alzheimer's disease (AD) neuropathology.1 Given the importance of these brain regions for sensorimotor integration and motor control, language function, emotional processing and social comportment, the principal clinical syndromes that result from FTLD, collectively known as frontotemporal dementia (FTD), are clinically diverse and severely disabling. The most common FTD syndrome is behavioural variant FTD (bvFTD), which is marked by early and severely disabling changes in personality and behaviour often occurring in the absence of cognitive impairment.
Historically, physicians recognised bvFTD under the narrow rubric of Pick's disease (PiD), and considered this disorder in the differential diagnosis of older adult patients presenting with dementia along with disinhibition and impulsivity.2 At the time, PiD could only be diagnosed definitively via autopsy. Presently, however, bvFTD is considered a common form of early-onset neurodegenerative disease, and the clinical and radiographic features of bvFTD have been delineated to allow premortem diagnosis.3 The term PiD is now reserved to identify a specific pathological subtype of FTLD. This expanded scope is recognised in Diagnostic and Statistical Manual of Mental Disorders (DSM) 5, where bvFTD has been designated as a neurocognitive disorder (NCD) due to FTLD pathology.
bvFTD is highly relevant to psychiatry as syndromic overlap exists between early bvFTD and several primary psychiatric disorders. bvFTD is marked by a wide range of changes in behaviour and personality that manifest long before cognitive changes ensue; these changes include psychosis, disinhibition, compulsions, overeating, loss of empathy and apathy. Therefore, individuals with bvFTD often present first in mental health settings where the differential diagnosis is challenging and the presence of early neurodegenerative disease may be missed altogether. Separating bvFTD from primary psychiatric disorders is important, as early accurate diagnosis helps prevent unnecessary clinical evaluations, allows timely discussions with caregivers about disease prognosis and management,4 and guides the institution of evidence-based supportive therapies.5 Furthermore, emerging disease-modifying therapies for FTLD are likely to be most effective in the earliest stages of the disease,6 and early accurate diagnosis facilitates early enrolment of patients in ongoing and future treatment trials.
Additionally, bvFTD provides a powerful paradigm for investigating the neural basis of primary psychiatric disorders. Whereas current diagnostic classification in psychiatry is based on identifying recognisable syndromes of observable behaviour, syndromic diagnoses in FTD are guided by knowledge of relevant functional neuroanatomy and neuropathology. Indeed, studies on the neural circuitry underlying bvFTD, and the neurodegenerative changes (ie, FTLD) affecting these circuits, are underway.7 Current trends in psychiatry are moving the field towards similar neural-based and biomarker-based approaches for diagnosis and treatment. The National Institute of Mental Health (NIMH) Strategic Plan includes explicit initiatives for integrating observable behaviour with biological markers, and developing—at least for research purposes—a diagnostic classification based on dimensions of behaviour and neurobiological mechanisms.8 Towards this end, bvFTD provides a rare model for moving our understanding of a psychiatric disorder from the syndromic level to a neural circuit-based model.
This article will first provide an overview of the FTD clinical syndromes, followed by a review of the molecular and histological changes observed in FTLD. Emphasis will be placed on bvFTD, which is the most common clinical syndrome associated with FTLD, and the most likely to be encountered in psychiatric settings. To illustrate how cognitive and behavioural symptoms are driven by neuroanatomy, this article provides a brief outline of the neural circuits that, when affected by FTLD, lead to dramatic behavioural, emotional and personality changes. We then review several case reports that illustrate the syndromic overlap that exists between bvFTD and several primary psychiatric disorders. Finally, we discuss future challenges in the field.
Frontotemporal dementia and related clinical syndromes
The focal onset of neurodegeneration in FTLD leads to distinct clinical syndromes early in the disease course. When the disease process is focused in the anterior frontotemporal regions, patients present with one of two clinical syndromes: (1) bvFTD, which is characterised by significant changes in emotion, behaviour, personality, and/or executive control3 or (2) primary progressive aphasia (PPA), marked by the slow deterioration of language function and/or semantic memory, usually in the absence of significant changes in comportment or impairment of other cognitive faculties.9 Collectively, bvFTD and PPA are referred to as frontotemporal dementia (FTD) in the literature; each of these syndromes will be described in greater detail below.
In a minority of cases, the neurodegenerative disease process is focused in cortices involved in motor control and sensorimotor integration, leading to clinical syndromes that are dramatically different from bvFTD and PPA, and include predominately motor syndromes such as corticobasal syndrome (CBS), progressive supranuclear palsy syndrome (PSP-S), and motor neuron disease (MND) syndromes.10 These syndromes have been described in other reviews and will not be discussed in this article.
In the late phases of FTLD, owing to the progressive spread of the neurodegenerative disease process along large-scale neural networks,7 patients with bvFTD often develop PPA, and patients with PPA often develop changes in behaviour and personality, and all patients with FTLD inevitably progress to levels of cognitive and motor impairment commensurate with global dementia.11
Primary progressive aphasia
The PPA syndrome is subdivided into three main clinical subtypes: the agrammatic (PPA-G), semantic (PPA-S) and logopenic (PPA-L) subtypes. PPA-L is most often caused by underlying AD neuropathology, and therefore will not be discussed further in this article. PPA-G and PPA-S are caused by FTLD neurodegeneration and will be discussed here briefly.
Each PPA subtype has well-defined diagnostic criteria based on clinical and radiographic features.9 In the earliest stages of disease, patients with PPA-G present with isolated difficulty maintaining grammatical constructs, often manifesting as improper use of verb tenses or adjectives. In other patients, non-fluent speech, decreased phrase length, effortful speech and dysprosody are evident. Brain imaging of these patients reveals predominant left posterior frontoinsular atrophy on MRI, and/or hypoperfusion/hypometabolism in this area on single-photon emission CT (SPECT)/positron emission tomography (PET). Patients with PPA-S experience an insidious loss of meaning of words, objects and other factual information about the world, while grammatical knowledge remains well preserved. These patients reveal predominant ATL atrophy on MRI and/or hypoperfusion/hypometabolism of this area on SPECT/PET imaging. Patients with PPA-S and PPA-G often have language-comprehension problems due to loss of word-meaning and impoverished grammar, respectively. Psychiatric manifestations occur in patients with PPA due to FTLD, particularly in PPA-S; however, psychiatric manifestations in PPA most often occur in the setting of a preceding or concurrent progressive language syndrome, which should alert clinicians to the presence of a neurological disease. This is not the case in bvFTD, which presents with isolated psychopathology.
Behavioural variant frontotemporal dementia
Patients with bvFTD experience dramatic changes in baseline personality and behavioural traits, which may initially occur in the absence of obvious cognitive impairment, as illustrated in the vignette below.3 These changes represent clinical manifestations of dysfunction in well-defined brain circuits that have reciprocal neural connections between the orbitofrontal, dorsolateral prefrontal and medial prefrontal cortices (see figures 1 and 2 for neuroanatomical orientation) and subcortical brain nuclei (basal ganglia and thalamic structures). These circuits are in turn influenced by cortical inputs from the temporal and parietal lobes.7 ,12 ,13 Normal function of these brain circuits correlates with normal social behaviours and emotional states in humans,12 ,13 and the gradual breakdown of these circuits due to FTLD leads to the bvFTD syndrome. Below, we briefly review the cardinal clinical signs and symptoms of bvFTD in light of relevant cortical functional neuroanatomy.
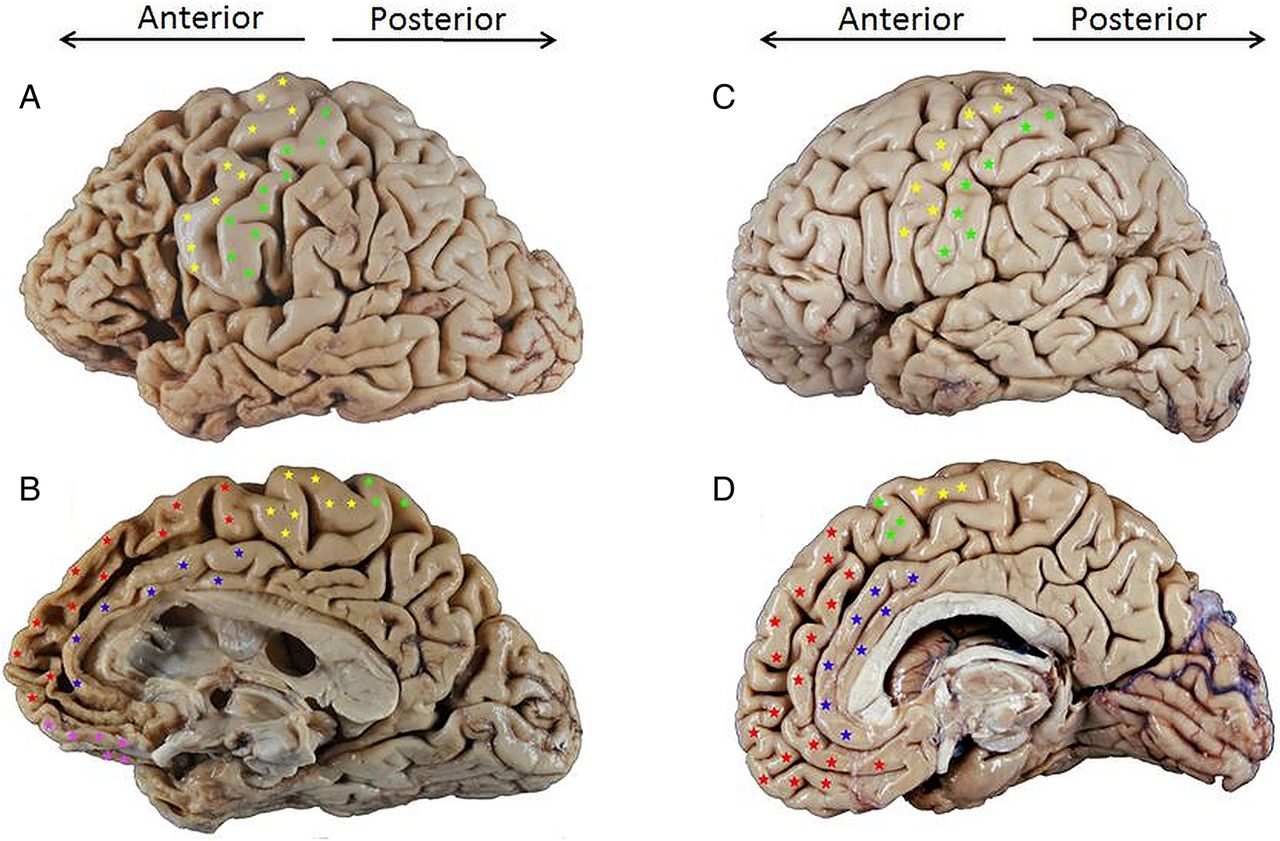
Lateral (A and C) and medial (B and D) views of the right hemisphere of a male patient with behavioural variant frontotemporal dementia (bvFTD) due to frontotemporal lobar degeneration (FTLD) τ-Pick's disease (A and B) compared to an age-matched normal brain (C and D) (A and C were flipped for easier comparison). The dorsolateral prefrontal cortex (DLPFC) is seen on the lateral surface of the brain, anterior to the primary motor and premotor cortices (indicated by yellow stars above) in the frontal lobe. In a normal brain (C), the volume of cortical gyri (folds) and the width of sulci (grooves) of the DLPFC are roughly equal to those of gyri and sulci in more posterior brain regions. In the affected brain (A) there is prominent atrophy (volume loss) of gyri within the DLPFC, leading to widening of sulci compared to more posterior brain regions; this atrophy pattern is striking when compared to the DLPFC of the normal brain (C). A similar degree of atrophy can be seen on the medial/mesial surface of the frontal lobe of the affected brain (B) compared to the normal brain (D); the affected brain shows prominent atrophy of the anterior cingulate gyrus (blue stars) and superior frontal gyrus (red stars), as well as the orbitofrontal cortex (pink stars).
{kind=link}
{kind=link}
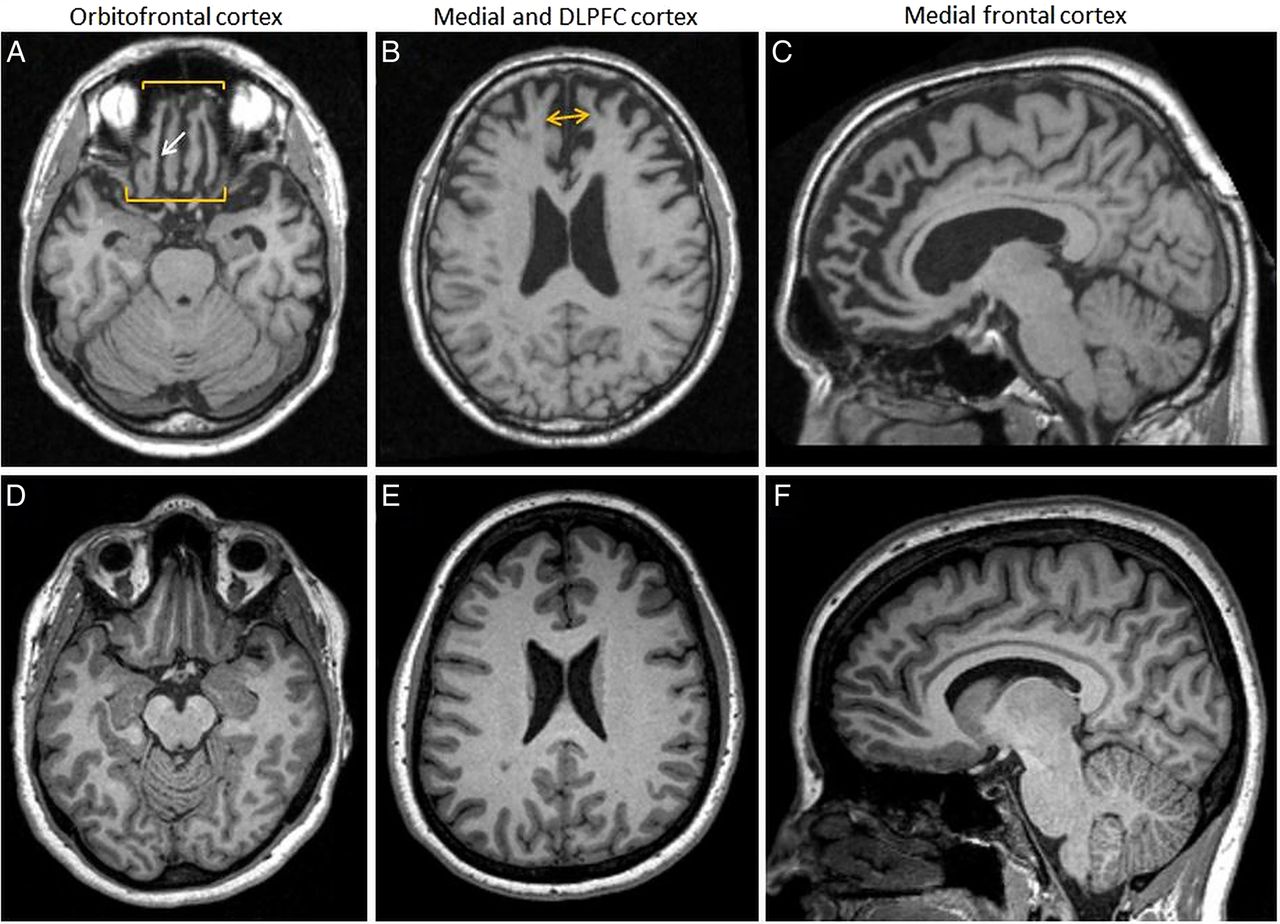
Brain MRI T1 of the patient presented in figure 2 7 years before death due to frontotemporal lobar degeneration (FTLD) τ-Pick's disease (A–C) compared to an age-matched normal control (D–F). There is prominent atrophy of the orbitofrontal cortex seen on axial T1 images (A); note prominent widening of the olfactory sulcus (A, white arrow) compared to normal (D). There is also prominent atrophy of the mesial frontal lobes seen on axial T1 images (B), as indicated by space between yellow arrowheads, compared to normal (E). On sagittal views, there is significant atrophy of the superior frontal gyrus and anterior cingulate gyrus (C) compared to normal (F).
Normal activity in the medial prefrontal cortex, including the anterior cingulate cortex (ACC), is strongly associated with motor and emotional motivation.13 ,14 Lesions in this area lead to dramatic reductions in spontaneous motor behaviours including speech. They also impede goal-directed behaviours in general, and produce marked psychomotor slowing. The ACC is most often attacked by the neurodegenerative disease process in bvFTD, particularly on the right side,15 and manifests clinically as marked apathy or inertia.3 Patients with apathetic bvFTD may spend all waking hours watching television, without attending to personal hygiene or other personal and social responsibilities. Patients with apathetic bvFTD do not initiate conversation and speak only when prompted, if at all. Apathy in bvFTD is often an early feature,3 but when it presents late in the disease it may ‘mask’ the more obvious signs of frontal lobe dysfunction. Clinically, there is notable overlap with the apathy associated with schizophrenia.
Patients with bvFTD often lose the ability to interpret and/or process the emotional states of people and animals. Thus, they demonstrate a general lack of self-awareness, and lack of empathy and sympathy towards strangers, friends and family, as well as pets. Spouses of patients with bvFTD often comment on their lack of interpersonal warmth, and diminished response to social cues. Such lack of empathy/sympathy in bvFTD is also predominately a right-sided manifestation of the disease including the right ATL, as well as the right medial orbitofrontal cortex (OFC) and anterior insula.16–19
Disinhibition is perhaps the most striking feature of bvFTD. The OFC may be conceptualised as a ‘relay station’ between higher order frontal cortices that inhibit a more ‘primitive’ (ie, reward-driven) limbic cortex; therefore, the OFC plays a critical role in modifying behaviour according to perceived reward and punishment values.13 ,20 Dysfunction of the OFC, particularly on the right side, is associated with behavioural disinhibition (ie, poor reward/punishment processing) and emotional lability. Right OFC neurodegeneration in bvFTD manifests as marked behavioural disinhibition,21 such as cursing, hugging or kissing strangers, urinating in public, performing inappropriate sexual acts, or telling offensive jokes to inappropriate audiences. Behavioural disinhibition may also manifest as impulsivity, such as reckless driving, new-onset gambling or substance abuse, excessive buying of unnecessary items, or criminal behaviours. Criminal behaviours occur in up to 54% of patients with bvFTD.22 Impulsivity related to food consumption is especially common in bvFTD, and this is also a manifestation of right OFC neurodegeneration, as well as insular, ventral striatal and ventral hypothalamic dysfunction.23 Patients with bvFTD tend to prefer sweet foods although some manifest indiscriminate hyperphagia; this results in significant weight gain.
Patients with bvFTD may engage in simple repetitive movements or verbal utterances, such as clapping, tapping, rubbing, humming and throat clearing, among others. More complex repetitive movements and utterances (that acquire a ritualistic or compulsive character) occur, such as repetitive counting, complex organising and cleaning routines, repetitive trips to the bathroom, or repetitive verbalisation of phrases or stories that have no communicative value. The brain localisation of these phenomena has not been studied satisfactorily in bvFTD, but there is evidence to suggest that frontosubcortical dysfunction is also responsible for these phenomena.24 From a phenomenological perspective, at our centre we do think of these repetitive phenomena as motor manifestations of disinhibition/impulsivity.
Finally, deficits in executive function, which are commonly seen in bvFTD, although not always early in the disease course, occur due to involvement of the dorsolateral prefrontal cortex.12 ,13 This manifests as trouble in multitasking, poor recall of recently learned material (poor working memory), mental rigidity, and environmental dependency (utilisation and imitation behaviours).3 These changes are not invariably present during the first phases of the illness, and can be found with many other conditions ranging from AD, parkinsonian dementias, cerebrovascular disease and depression.
It is important to note that the signs and symptoms of bvFTD can all occur in a single patient at different time points in the disease course. This is presumably the result of the spatial and temporal spread of the neurodegenerative disease process although further staging clinicopathological studies are needed to substantiate this hypothesis.
bvFTD case vignette: The following vignette briefly summarises the most salient clinical features of a patient with autopsy-confirmed bvFTD who was diagnosed and followed in our centre.
The patient was 55 years of age when she first came to our attention. She was right-handed. Her doctor referred her to our centre at the insistence of the patient's husband, who upon learning of bvFTD thought his wife may have the disease. The first serious ‘red flag’ occurred during a family trip abroad 4 years prior to presentation; she made several racial comments about a restaurant server despite clearly causing discomfort to everyone present, including the server. A few months later, she repeatedly interrupted her son's graduation ceremony by initiating conversation with guests during the ceremony. She also attempted to take a bouquet of flowers from another family to give it to her son. Her behaviour in social situations became increasingly erratic over the ensuing 2 years. She regularly initiated conversations with strangers about sex, and also made sexual jokes in front of children. She worked in an elementary school at the time, and her supervisor noted that she developed a tendency to play roughly with children, to the extent that she made some of them cry. She lost her job 3 years after onset of her illness. One year before seeing us, she developed a voracious appetite for sweets leading to a 25-pound weight gain within 12 months. By the time we first saw her in our centre, her behaviours had diminished in intensity. Her neurological examination was notable for stimulus-bound behaviours and lack of respect for social boundaries. She repeatedly tried to kiss and hug the examiner. There were no signs of MND. Neuropsychological testing was notable for a score of 26/30 on the mini-mental state examination. She was presented with 16 photographs of faces with a particular emotion, and was able to correctly identify the emotion of only 6 faces (normal=13.3±1.7). On the Interpersonal Reactivity Index—Perspective Taking (IRI-PS), she scored 8/35 (normal=24.5±5.5), suggestive of a profound inability to think of others’ emotions; likewise, she scored 15/35 (normal=28.6±4.5) on the IRI empathic concern (IRI-EC) scale, suggestive of a profound impairment in her ability to experience the emotional state of others. Her brain MRI revealed marked bilateral frontal and anterior temporal lobar atrophy. Genetic testing revealed a C9ORF72 gene expansion (interestingly, and pertinent to this review article, the patient's family history was only notable for a distant paternal uncle with suspected Parkinson's disease). Her syndrome evolved into a predominantly apathetic state. She died at age 59 years, approximately 8 years after onset. Brain autopsy confirmed FTLD.
Epidemiology of FTD
FTD is one of the leading causes of early-onset dementia, which is somewhat arbitrarily defined as dementia occurring before the age of 65 years.25 FTD may occur as early as the second decade of life,26 ,27 and 13% of FTD cases occur in people younger than 50 years, according to population-based studies.28 The mean age of onset is approximately 56 years. Nearly 40% of cases are familial, and 10–15% FTD cases appear to be caused by an autosomal dominant pattern of inheritance. The estimated combined population prevalence of FTD in individuals 45–64 years of age, according to studies conducted in North America, Asia and Europe, is 2–31/100 000,28 which is similar to that of early-onset AD (45–64 years of age) in European and North American populations (15–44/100 000).29
Data on the incidence of FTD is largely based on three population studies conducted in Europe and the USA,28 which revealed an incidence of 2.7–4.1/100 000 in individuals <70 years. When broken down by age, the incidence of FTD for the age groups 40–49, 50–59 and 60–69 years were, respectively, 2.2, 3.3 and 8.9/100 000 person-years in Rochester, USA, and 1.2, 2.4 and 7.7/100 000 person-years in Girona, Spain.28 Some studies suggest that the incidence of FTD is higher than AD among individuals <60 years of age.30
Clinically, bvFTD is the manifesting syndrome of FTLD in 50–57% of autopsy-confirmed cases, whereas roughly 40% of autopsy-confirmed FTLD cases manifest as PPA.10 ,31 The primary motor manifestations of FTLD (PSP-S, CBS, MND) are seen as the initial symptoms in less than 5% of cases. Estimates of disease duration in FTD vary widely. According to specialist clinic cohorts, survival varies from 3 to 14 years,28 although a well-recognised subset of patients manifest a very slowly progressive form of FTD,32 and there are individual cases of disease duration lasting more than 20 years.33
FTLD neuropathology: distinct abnormal protein aggregates and causative genetic mutations
Macroscopic and microscopic characteristics
Macroscopically, FTLD results in varying degrees of focal atrophy of all the cortical regions that form part of the cortical-subcortical brain circuits discussed above. These include limbic, paralimbic and association cortices of the frontal and temporal lobes, insular cortex, as well as atrophy of subcortical nuclei (basal ganglia and thalamic structures).34 Brain atrophy due to FTLD is observed on gross brain sections (see figure 1), and can also be appreciated on brain neuroimaging (see figure 2). Microscopically, all forms of FTLD demonstrate neuronal loss, manifesting as tissue vacuolation, and inflammatory changes, or gliosis, of the affected regions (see online supplementary figures A and B).34
Abnormal protein inclusions
At a molecular level, nearly all cases of FTLD are associated with the accumulation of one of four abnormal protein inclusions (or proteinopathies) in neurons and/or glial cells of the brain, which serve to divide FTLD into four main diagnostic categories (see online supplementary figures C–F):35 ,36 (1) τ protein (FTLD-τ), (2) transactive response DNA-binding protein 43 (FTLD-TDP), (3) fused-in sarcoma (FTLD-FUS) protein, and (4) dipeptide proteins generated from mutant forms of the C9ORF72 gene (see genetics section below).
FTLD is nearly evenly divided between FTLD-τ and FTLD-TDP, whereas about 10% of cases are associated with FUS and other substrates including C9ORF72 dipeptides.1 FTLD-τ and FTLD-TDP-43 are further subdivided into several subtypes, based on specific morphological features, biochemical properties, and distribution of protein aggregates within the brain. For example, the diagnostic subtypes of FTLD-τ include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and PiD. Note that these designations are used independently of similar designations used to identify specific clinical syndromes. For example, although most cases of CBS are caused by CBD, CBS can also be caused by PSP and even AD pathology.
Causative genetic mutations
From 30% to 50% of FTD cases are familial, and up to 10–15% show an autosomal dominant pattern of inheritance. Presently, mutations in three genes have been linked to the majority of familial FTLD,37 ,38 with nearly 100% penetrance:28 microtubule-associated protein τ, granulin (GRN), and C9 open reading frame 72 (C9ORF72). To date, the largest proportion of familial FTD (12–25%) is caused by pathogenic expansions of C9ORF72.28
Five other genes have been linked to a few families affected with FTLD: valosin-containing protein, charged multivesicular body protein 2b, fused-in sarcoma (FUS), TAR DNA binding protein, and the UBQLN2 gene. These genes and their associated phenotypes will not be discussed further in this article.
Clinical, pathological and genetic correlations in FTLD
The recent discovery of FTLD-causative genes has led to a convergence of knowledge around the neuropathology, neuroimaging and clinical syndromes that continues to expand our appreciation of hereditary and sporadic FTLD-associated phenotypes, beyond bvFTD and PPA, as shown in table 1. Moreover, all the clinicopathological correlations identified in hereditary FTLD shown in table 1 have also been found to occur in sporadic FTLD.10
Clinicopathological correlations in hereditary frontotemporal lobar degeneration (FTLD)
Given the wide variety of functions ascribed to the human frontal and temporal lobes, it is perhaps not surprising that sporadic and hereditary forms of FTLD lead to such a varied spectrum of clinical manifestations.10 This clinical heterogeneity, and the fact that different FTLD subtypes (τ, TDP, FUS and C9ORF72-associated dipeptides) lead to similar clinical syndromes, may be interpreted as empirical proof that the distribution and spread of pathology in the brain, and perhaps not so much the type of pathology, dictates a patient's clinical syndrome. Indeed, although physiological dysfunction,43 gliotic changes and synaptic loss34 are collectively responsible for clinical decline in FTLD, specific syndromes result from involvement of distinct brain circuits.
Diagnosing bvFTD
Diagnostic criteria
The International behavioural Variant FTD Criteria Consortium (Frontotemporal Dementia Consortium, FTDC) published consensus criteria for bvFTD in 2011.3 These criteria are rooted on the clinical, neuroanatomical and neuropathological data described above, and they represent the diagnostic gold standard for bvFTD. These criteria stratify the bvFTD diagnosis hierarchically into three categories—possible, probable and definite—based on increasing levels of certainty attained according to clinical, neuroimaging, molecular and histopathological data, as shown in table 2. In short, whereas possible bvFTD is diagnosed based exclusively on clinical signs and symptoms, probable and definite bvFTD are supported by radiological findings and neuropathological data (brain tissue or genetics), respectively. These criteria were shown to be superior to previously published criteria for bvFTD.44
Frontotemporal Dementia Consortium (FTDC) and Diagnostic and Statistical Manual of Mental Disorders (DSM-5) criterion sets for behavioural variant frontotemporal dementia (bvFTD) and mild or major behavioural variant Frontotemporal Neurocognitive Disorder (bvFTNCD), respectively3
The fifth edition of the DSM-5 also contains diagnostic criteria for bvFTD (shown in table 2), within a new classification system for cognitive disorders. Briefly, in the DSM-5, all acquired primary cognitive disorders are referred to as NCDs. The DSM-5 recognises that many patients with chronic psychiatric disorders exhibit cognitive impairment; hence, all patients with NCD must exhibit cognitive impairment as a core feature of their respective syndrome. Accordingly, cognitive impairment is defined by the presence of subjective and objective signs/symptoms in at least one of six neurocognitive domains: complex attention, executive function, learning and memory, language, perceptual motor abilities and social cognition. NCDs are further subdivided into the syndromes of Mild and Major NCD, according to severity of cognitive and functional impairment. Specific diagnostic criteria are subsequently provided for 13 aetiological subtypes of Mild and Major NCD, including Mild and Major Frontotemporal NCD. Table 2 presents both the FTDC and DSM-5 bvFTD criterion sets next to each other for comparison. Important similarities and differences between these criteria will be discussed later in this article.
Previous editions of the DSM do not include social cognition among the more traditional brain-based cognitive domains (memory, language, etc). Hence, it is important to discuss this domain briefly, particularly given its involvement in bvFTD. Broadly, social cognition is defined as ‘the processes by which people understand themselves and other people’.45 The principal dimensions of social cognition include self-perception (ie, awareness of one's own emotional and mental processes in response to internal and/or external stimuli) and person-perception (ie, awareness of other people's feelings and emotions in specific contexts, also referred to as ‘theory of mind’), as well as knowledge of social norms and expectations (ie, ethical and moral behaviour).45 It has long been recognised that all dimensions of social cognition are entirely dependent on specific regions/structures of the frontal and temporal lobes of the brain (predominately on the right).45 Indeed, the seamless integration of these neural processes is necessary for normal social behaviour. As can be gathered from the previous neuroanatomical discussion of the signs and symptoms of bvFTD and the associated case vignette, the principal dimensions of social cognition are typically severely disrupted in patients with bvFTD owing to the fact that the neurodegenerative process in these patients is centred in the frontal and temporal lobes.
Differential diagnosis of early bvFTD
By the time patients with bvFTD are referred to a neurologist, patients are usually in the middle stages of the disease, when the differential diagnosis is relatively narrow, and accurate syndromic and neuropathological diagnosis is achieved with high sensitivity and specificity based on clinical, neuropsychological, radiographic, genetic and cerebrospinal fluid (CSF) biomarker data (CSF analysis is useful to rule in AD and to rule in/out degenerative/non-degenerative causes of rapidly progressive dementia).46
The differential diagnosis of early bvFTD is far more challenging largely due to the syndromic overlap that exists between early bvFTD and late-onset psychiatric disease. The psychopathological changes of early bvFTD (disinhibition, impulsivity, apathy, compulsive behaviours, etc) can occur in the absence of other neurological signs or symptoms, including common signs of cognitive impairment such as memory loss.3 The paucity of neurological and/or cognitive complaints, and the lack of insight observed in early bvFTD,47 leads caregivers and primary physicians to routinely seek psychiatric evaluation for such patients. Therefore, non-specific/atypical psychiatric presentations have long been recognised to occur in early bvFTD,48–50 dating back to the 1940s when bvFTD was referred to as PiD.51 Furthermore, in a significant proportion of patients, the psychopathology of early bvFTD may cluster into DSM-compatible psychiatric diagnoses. Indeed, in one large study, up to 50% of patients diagnosed with bvFTD received a previous psychiatric diagnosis per DSM criteria.52 A sample of these and other cases is presented below.
bvFTD manifesting as suspected primary psychiatric disorders
Early bvFTD psychiatric manifestations include psychotic, mood and obsessive–compulsive disorders. Table 3 shows a representative sample of the various DSM-compatible psychiatric diagnoses assigned to patients who were subsequently diagnosed with possible, probable, or definitive bvFTD.
Sample of DSM-compatible diagnoses assigned to patients who were subsequently found to have possible, probable and definitive bvFTD
Psychotic, bipolar and obsessive–compulsive disorders have been reported as initial manifestations of bvFTD for decades. According to a review of 751 selected cases of FTD published between 1950 and 2007, 6% (46 cases) presented with schizophrenia, schizoaffective disorder, bipolar disorder (BPD), psychotic depression, or unspecified psychotic states, based on clinical, neuropathological and/or genetic criteria available at the time of the publication of each case.63 Also, obsessive–compulsive behaviours have also long been recognised as initial manifestations of bvFTD;51 disease manifestations included compulsive counting of objects, door-locking and hand washing, which were considered indistinguishable from ‘obsessive neurosis’.51
The gamut of psychiatric manifestations in early-stage bvFTD has expanded with the identification of FTLD due to C9ORF72 (see table 3 and online supplementary table). Carriers of C9ORF72 mutations are especially prone to psychiatric manifestations at disease onset,39 ,64 ,65 mainly presenting as psychotic, bipolar and compulsive disorders. For example, in a recent FTD study that included 29 patients with bvFTD due to C9ORF72 mutations, 12 patients (41%) exhibited psychosis as a dominant presenting problem, resulting in initial diagnoses of delusional psychosis, somatoform psychosis, or paranoid schizophrenia.39 The mean duration of symptoms at the time of referral to the dementia clinic was 2.7 years (SD=2.4). The online supplementary table presents some of the initial psychiatric manifestations in this patient cohort. Conversely, screening of C9ORF72 gene expansions in a large sample of subjects with BPD revealed a proband with BPD and a C9ORF72 gene expansion, whose father also carried a C9ORF72 expansion and died of autopsy-confirmed FLTD that initially manifested as BPD.66
Psychiatric manifestations were also common in an analysis of 32 patients with FTD due to GRN mutations, including bulimia, personality changes, sexual disinhibition, ritualistic behaviours and paranoia;67 8 patients in this cohort (25%) manifested visual hallucinations, 1 manifested both auditory and visual hallucinations, and 2 members of a GRN mutation-carrying family carried a diagnosis of schizophrenia.67 Psychotic68 and bipolar50 syndromes have been associated with GRN mutations, and recent studies indicate that different polymorphisms of GRN may contribute to schizophrenia and BPD, thus suggesting the possibility of shared pathophysiological mechanisms between FTD and some psychotic disorders.69 This notion is further supported by the fact that plasma levels of progranulin were found to be lower in patients with BPD (without GRN mutations) compared with controls in two different study populations.69 ,70
It is important to mention that we have focused our review on patients who received a DSM-compatible primary psychiatric diagnosis and were later found to have bvFTD, therefore suggesting that the psychopathological changes that led to a primary psychiatric diagnosis were actually the earliest clinical manifestations of FTLD. We do not intend to imply that a primary psychiatric diagnosis and bvFTD cannot co-occur in a patient, although dual diagnoses are difficult to establish with certainty at present mainly due to the lack of well-established biomarkers available to ‘rule in’ primary psychiatric disorders. Therefore, in our opinion, and based on the cases reviewed above, clinicians evaluating adult patients with ‘late-onset’ or ‘atypical’ psychiatric disease should consider early bvFTD in their differential diagnosis, while being fully aware that primary psychiatric disorders are far more common than bvFTD.
The bvFTD phenocopy syndrome and slowly progressive FTLD
There is a well-documented subset of patients who manifest typical signs and symptoms of bvFTD yet do not show clear biomarker evidence of FTLD, and they do not clearly demonstrate progressive clinical deterioration (hence they do not meet diagnostic criteria for bvFTD).32 ,71 ,72 These patients are described as bvFTD phenocopy syndrome (bvFTD-PS) in the literature.32 The clinical overlap between bvFTD-PS and other neuropsychiatric conditions has been noted,32 and in view of the preceding discussion of the syndromic overlap between early bvFTD and several primary psychiatric disorders, we suspect many patients with bvFTD-PS actually suffer from a primary psychiatric disorder, and vice-versa. Along this line of thought, the opportunity to diagnose definite bvFTD via genetic testing has allowed the recognition of very slowly progressive forms of bvFTD, with disease duration upwards of 20 years.33 We therefore also suspect that cases of slowly progressive bvFTD may be particularly difficult to differentiate from patients with a primary psychiatric disorder.
Discussion
In this article, we have shown that the psychopathological manifestations (ie, changes in personality and behaviour) of early bvFTD frequently mimic those seen in DSM-compatible primary psychiatric disorders. Since the classification of psychiatric diagnoses, as exemplified by the DSM-5, relies exclusively on clinical phenotypic profiles, and there is considerable phenotypic overlap between patients with early bvFTD and age-matched patients with primary psychiatric disorders; patients with bvFTD are often misdiagnosed and managed as psychiatric patients for years before being accurately diagnosed with a neurodegenerative disease.
Several factors may be contributing to misdiagnosis. Lack of well-established biomarkers for psychiatric disorders is one of them, as presently there is no objective way of ‘ruling in’ psychiatric disease. On the other hand, we suspect that psychiatrists do not regularly make use of well-established bvFTD biomarkers (ie, MRI, PET, SPECT and genetics) when evaluating these patients. Use of these bvFTD biomarkers in psychiatric settings is important, since the individual psychotic manifestations of bvFTD due to a C9ORF72 mutation (ie, hallucinations and delusions), for example, can be clinically indistinguishable from those seen in idiopathic schizophrenia.
Additionally, two features of the DSM-5 diagnostic criteria for bvFTD may inadvertently discourage the early recognition of this disease in mental health settings (see table 2). First, by requiring cognitive impairment as a core feature of bvFTD, the condition may be missed, as marked cognitive changes are rare in the earliest stages of disease. Although the DSM-5 emphasises that the required cognitive deficit may be in the domain of social cognition, we suspect this domain is not well recognised as a brain-based cognitive domain in most clinical settings, compared with other more traditional cognitive domains such as memory or language function. Besides, impaired social cognition is seen in many other primary psychiatric disorders,73 hence, the presence of impaired social cognition per se may not prompt a psychiatrist to consider bvFTD in the differential diagnosis of patients with prominent changes in behavioural and personality. Second, while both bvFTD criterion sets stratify the diagnosis into possible and probable categories based on analogous clinical and/or neuroimaging data, the DSM-5 does not include a definite bvFTD diagnostic category, and regards the presence of an FTLD-causative genetic mutation as evidence of probable bvFTD. In practice, this difference may lead psychiatrists and other physicians to conclude that a bvFTD cannot be diagnosed definitively during life (hence, workup is not pursued), when in fact definite bvFTD can be diagnosed via genetic testing. Indeed, these mutations are considered to be 100% penetrant,28 meaning, that all mutation carriers will develop FTLD if they live long enough to express the disease. In practice, this means that in a subset of patients, clinicians can diagnose bvFTD definitively in a patient with possible bvFTD (ie, positive clinical syndrome in the absence of brain imaging abnormalities) via genetic testing, although further research is needed to identify the specific profile of patients with psychiatric illness in whom genetic testing for FTLD may be useful and cost effective in the clinical setting.
Accurate early diagnosis of bvFTD is important for several reasons: (1) it leads to timely caregiver education and family counselling regarding neurodegenerative disease, as well as early institution of evidence-based supportive therapies for bvFTD;74 (2) early diagnosis prepares caregivers for the psychological and economic demands of caring for a loved one with neurodegenerative disease, and helps delay institutionalisation;4 and perhaps most importantly, (3) disease-modifying therapies for bvFTD, which are already under development, will be most effective in the earliest stages of disease.6 Once disease-modifying therapies for bvFTD become available, genetic testing will become an invaluable tool to help clinicians separate early bvFTD from primary psychiatric disorders in selected groups of patients, and refer these patients for treatment. Presently, aside from family history, the clinical profiles of psychiatric patients who are most likely to carry an FTLD-causative genetic mutation are unknown, and this is an area that deserves the attention of clinical research.
Towards the goal of recognising bvFTD in the earliest stages, we strongly recommend that clinicians not only consider bvFTD in the differential diagnosis of dementing disorders like AD, but also in the differential diagnosis of conditions such as schizophrenia and BPD and any psychiatric disorder in patients with known familial forms of dementia. The average practitioner's repertoire of clinical skills should be enhanced to systematically examine social cognition, and consider it as a cognitive domain together with other more traditional cognitive domains such as language and memory. In mental health settings where it is difficult to order neuroimaging procedures, practitioners should make themselves aware of colleagues in psychiatry or neurology who are experts in dealing with neurodegenerative disorders and to whom patients can be referred for consultation. Psychiatrists should become aware of resources for their patients and families such as The Association for Frontotemporal Degeneration (http://www.theaftd.org), and of opportunities for their patients to participate in clinical trials of disease-modifying therapies for FTLD.
From a neuroscience perspective, we hope that the clinical phenotypic overlap between early bvFTD and primary psychiatric disorders generates academic interest in the exploration of bvFTD (and other neurodegenerative diseases of the brain) as models to understand the neural basis of the behavioural and emotional symptomatology of primary psychiatric disorders.75 As discussed in this article, bvFTD provides empiric proof that the distribution of pathology in the brain along distinct brain circuits dictates the clinical phenotype. Although the neuropathological substrate/s of primary psychiatric disorders remain elusive, the frontal and temporal lobes, and their underlying neural circuits, are prime territory for investigations into psychiatric disease. Indeed, the overlap between the fields of neurology and psychiatry is most evident in disorders that affect the anterior frontotemporal regions and their subcortical circuits.
In conclusion, in this article we have reviewed the clinical manifestations of FTLD (ie, FTD and related syndromes), while focusing on bvFTD, which is the most common FTD syndrome and also the most commonly encountered in psychiatric settings. Owing to the variety of psychiatric manifestations that occur in early bvFTD, often in the absence of cognitive impairment, distinguishing early bvFTD from some primary psychiatric disorders is challenging. We have postulated possible contributors to this diagnostic challenge, and we have pointed out areas of basic and clinical research that deserve further attention.
Acknowledgments
The authors are grateful to Dr Mary Ganguli for contributions provided in the preparation of this article.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online figure
- Data supplement 2 - Online table
Footnotes
Funding SCL is supported by an NIH T32 grant. BLM receives grant support from the NIH/NIA (grants P50AG023501, P01AG019724, and P50 AG1657303). BLM also receives royalties from Cambridge University Press, Guilford Publications, Inc., and Neurocase.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.